プリオン病/亜急性硬化性全脳炎(SSPE)/進行性多巣性白質脳症(PML)とは
プリオン病とは
志賀裕正(東北大学附属病院神経内科)
プリオン病は脳内に存在する正常型プリオン蛋白が通常の方法では分解されない異常型プリオン蛋白に変換され蓄積し、神経細胞を障害することによって発病する進行性・致死性脳症です。また人獣に共通する感染性疾患でもあります。現在治療の試みが行われていますが、まだ根本的治療法は確立していません。
原因不明の孤発性Creutzfeldt-Jakob 病 (CJD)、プリオン蛋白遺伝子変異により発病する遺伝性プリオン病 (遺伝性といっても必ずしも家族歴があるとは限らず、家族集積性がなくあたかも孤発例のように発症する場合もあります)、感染原因のはっきりしている感染性CJD
(牛海綿状脳症と関連する変異型CJDや硬膜移植手術後に発症するCJD、パプアニューギニアで発生したKuru病など)に分類されます。日本ではプリオン病の約80%は孤発性CJDで、10%が遺伝性プリオン病、残りが感染性CJD
(1例が変異型CJDで残りは硬膜移植後CJD)となっています。
孤発性CJDは認知障害や視覚障害、小脳失調で発症することが多く、その他高次機能障害、錐体路および錐体外路徴候、ミオクローヌスなどが複合して出現、進行し平均して発症3.5ヶ月で無動無言に陥ります。初発症状や進行過程は多様で、早期診断が困難な場合も少なくありません。
検査所見では周期性同期性放電 (PSD)といわれる脳波異常(図1)が特異的で、髄液中に神経細胞特異蛋白といわれるNeuron Specific Enplase
(>25 ng/ml)や14-3-3蛋白、tau蛋白(>1,300 pg/ml)を検出することも比較的特異度の高い検査とされています。最近では拡散強調MRI
(DWI)が早期から異常を検出することが報告されています(図2)。急速に進行する精神・神経症状とこれらの検査をもとに診断を行います。
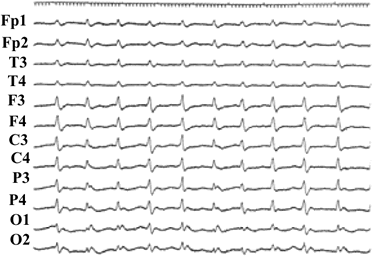
図1:CJDで認められる典型的なPSD
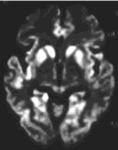
図2:孤発性CJD患者のDWI大脳皮質、基底核に高信号を認める。